競合阻害(きょうごうそがい、英: competitive inhibition)、競争阻害、拮抗阻害は、酵素の活性部位への阻害剤の結合が基質の結合を妨げる(逆もまた同様)酵素阻害剤の形式である[1][2]。
ほとんどの競合阻害剤は酵素の活性部位に可逆的に結合することによって機能する[1]。その結果、多くの文献ではこれが競合阻害剤を決定付ける特徴であると述べられている[3][4] 。しかしながら、酵素が阻害剤あるいは基質のどちらとも結合できるが同時には結合できない多くの可能な機構が存在するため、これは誤解を招くおそれのある過度の単純化である[1]。例えば、アロステリック阻害剤は競合的、非競合的、不競合的阻害を見せる可能性がある[1]。
機構
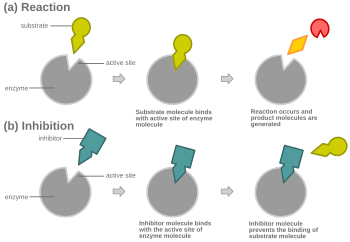 競合阻害を示す概略図
競合阻害を示す概略図 競合阻害において、いかなる時でも、酵素は阻害剤に結合した状態、基質に結合した状態、どちらとも結合していない状態をとれるが、阻害剤と基質の両方に同時に結合することはできない。
実質的に全ての場合において、競合阻害剤は基質と同じ結合部位へ結合するが、同一部位への結合は必要条件ではない。競合阻害剤は、基質が結合している時にアロステリック部位へ結合しない限りは、遊離酵素のアロステリック部位へ結合し、基質の結合を妨げることもありうる。
競合阻害では、反応の最大速度 ( ) は変化しないが、結合部位への基質の見かけの親和性が低下する(解離定数
) は変化しないが、結合部位への基質の見かけの親和性が低下する(解離定数 は一見増加する)。
は一見増加する)。 (ミカエリス・メンテン定数)の変化はの変化と平行である。いかなる競合阻害剤濃度も、基質が酵素への結合において阻害剤を打ち負かす場合では、基質の濃度を増加させることによって乗り越えることができる。
(ミカエリス・メンテン定数)の変化はの変化と平行である。いかなる競合阻害剤濃度も、基質が酵素への結合において阻害剤を打ち負かす場合では、基質の濃度を増加させることによって乗り越えることができる。
 阻害剤および基質が同時に酵素に結合できない限りは、競合阻害はアロステリックにもなりうる。
阻害剤および基質が同時に酵素に結合できない限りは、競合阻害はアロステリックにもなりうる。 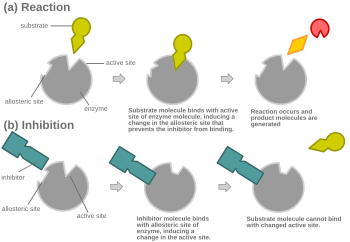 アロステリック競合阻害のもう一つの可能な機構。
アロステリック競合阻害のもう一つの可能な機構。 式
競合阻害はミカエリス・メンテン定数 の見かけの値を増加させる。反応初速度
の見かけの値を増加させる。反応初速度 は以下の式で与えられる。
は以下の式で与えられる。
![{\displaystyle V_{0}={\frac {V_{\max }\,[S]}{K_{m}^{\text{app}}+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7e6ea985b759c13c54c08b2e1a1f7b292c549beb)
上式において![{\displaystyle K_{m}^{\text{app}}=K_{m}(1+[I]/K_{i})}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e013bb3d416acd07c0c55ee196d46e9ede190a63) であり、
であり、 は阻害剤の解離定数、
は阻害剤の解離定数、![{\displaystyle [I]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5962300a54e8ce8b5761dac9a5fbbca450c2ce0f) は阻害剤濃度である。
は阻害剤濃度である。
阻害剤の存在はより高濃度の基質を用いることによって克服できるため、は変化しない。 に達するために必要な基質濃度であるは競合阻害剤存在下で増大する。これは、阻害剤存在時にに達するために必要な基質の濃度が阻害剤非存在時にに達するために必要な基質の濃度よりも大きいためである。
に達するために必要な基質濃度であるは競合阻害剤存在下で増大する。これは、阻害剤存在時にに達するために必要な基質の濃度が阻害剤非存在時にに達するために必要な基質の濃度よりも大きいためである。
導出
単一基質酵素がミカエリス・メンテン反応速度論に従う最も単純な場合における典型的なスキームである

は、遊離酵素への阻害剤の結合を含めて修正される。

ここで留意すべきは、阻害剤はES複合体へ結合せず、基質はEI複合体に結合しないことである。この挙動は両方の化合物が同じ部位へ結合していることを示すものであると一般的に見なされているが、これは厳密に必須ではない。ミカエリス・メンテン式の導出と同様に、系は定常状態、すなわちそれぞれの酵素種の濃度は変化しない仮定する。
![{\displaystyle {\frac {d[E]}{dt}}={\frac {d[ES]}{dt}}={\frac {d[EI]}{dt}}=0.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/268777cfb0aa902787812bbb71d192f24b85464d)
さらに、既知の酵素総濃度は![{\displaystyle [E]_{0}=[E]+[ES]+[EI]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/97e7fcec01c7884f769ae9c075e6d1a4911f42d7) 、反応速度は基質および阻害剤濃度が実質的に変化しない条件下で測定され、わずかな量の生成物が蓄積しているとする。
、反応速度は基質および阻害剤濃度が実質的に変化しない条件下で測定され、わずかな量の生成物が蓄積しているとする。
したがって、系について以下の式が成り立つ。
![{\displaystyle [E]_{0}=[E]+[ES]+[EI]\,\!}](https://wikimedia.org/api/rest_v1/media/math/render/svg/978d6a7643aa47348cfce3f507afb7841b127048)
(1)
![{\displaystyle {\frac {d[E]}{dt}}=0=-k_{1}[E][S]+k_{-1}[ES]+k_{2}[ES]-k_{3}[E][I]+k_{-3}[EI]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9965a57f03834ac7b5cde88df11e71be46057435)
(2)
![{\displaystyle {\frac {d[ES]}{dt}}=0=k_{1}[E][S]-k_{-1}[ES]-k_{2}[ES]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/51adba7afebf52de85559de2f95bc1ee080094a0)
(3)
![{\displaystyle {\frac {d[EI]}{dt}}=0=k_{3}[E][I]-k_{-3}[EI]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3c24cc7debf78e40ee442845a6bb0e8cc078aadf)
(4)
上式において![{\displaystyle [S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/292bbb82029aa583c5d2ac5fa1d7e4fedf537d8b) 、、
、、![{\displaystyle [E]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/409eb69aba4c3afa67a48af6d9f976a28445c544) は既知である。反応初速度は
は既知である。反応初速度は![{\displaystyle V_{0}=d[P]/dt=k_{2}[ES]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/46255eb4443a618d59991823c0c26a019eb1422e) と定義されるため、未知の
と定義されるため、未知の![{\displaystyle [ES]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/31639ab6b9c7c728139b5f8ce03991d800ac7741) を既知の、、の観点から定義する必要がある。
を既知の、、の観点から定義する必要がある。
式 (3) から、
![{\displaystyle k_{1}[E][S]=(k_{-1}+k_{2})[ES]\,\!}](https://wikimedia.org/api/rest_v1/media/math/render/svg/81f920641b7d4ee0c9c57a53ebd2a43cca9cea0a)
を変形することによって、EをESを単位として定義することができる。
両辺を![{\displaystyle k_{1}[S]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8f2a483dc97db9519f165c7f706ef334bddac55b) で割ると、
で割ると、
![{\displaystyle [E]={\frac {(k_{-1}+k_{2})[ES]}{k_{1}[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/daeace436c65cee7869e60b931f05cb1de334232)
となる。
ミカエリス・メンテン式の導出と同様に、 項は巨視的速度定数で置き換えることができる。
項は巨視的速度定数で置き換えることができる。
![{\displaystyle [E]={\frac {K_{m}[ES]}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d28bfe1a299584c50be6bff337c20855c3567a52)
(5)
式 (5) を式 (4) へ置換すると、
![{\displaystyle 0={\frac {k_{3}[I]K_{m}[ES]}{[S]}}-k_{-3}[EI]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/d54314972d85eabd9bb831f44a0737f4f7123e69)
変形すると、
![{\displaystyle [EI]={\frac {K_{m}k_{3}[I][ES]}{k_{-3}[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/668346ee94b193f238b09e941bcfe0a7ccc0fcfb)
となる。
この時点で、 として阻害剤の解離定数を定義することができ、
として阻害剤の解離定数を定義することができ、
![{\displaystyle [EI]={\frac {K_{m}[I][ES]}{K_{i}[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6fa68e2d01ebf1553c7e6c30996f3509b84ac1cb)
(6)
となる。
この時点で、式 (5) と式 (6) を式 (1) へ置換する。
![{\displaystyle [E]_{0}={\frac {K_{m}[ES]}{[S]}}+[ES]+{\frac {K_{m}[I][ES]}{K_{i}[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7b2c23abf3e9cc29f2d34091e793b1b10fbcdde6)
ESについて式を解くと以下の式が得られる。
![{\displaystyle [E]_{0}=[ES]\left({\frac {K_{m}}{[S]}}+1+{\frac {K_{m}[I]}{K_{i}[S]}}\right)=[ES]{\frac {K_{m}K_{i}+K_{i}[S]+K_{m}[I]}{K_{i}[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/195800a9b1300f606542d148f00b7e988ad3e2fa)
![{\displaystyle [ES]={\frac {K_{i}[S][E]_{0}}{K_{m}K_{i}+K_{i}[S]+K_{m}[I]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ecd2ed9ccebf668a3c52cb85c33195dad2bf6ded)
(7)
したがって、は
![{\displaystyle V_{0}=k_{2}[ES]={\frac {k_{2}K_{i}[S][E]_{0}}{K_{m}K_{i}+K_{i}[S]+K_{m}[I]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/55aaeac269215ee73c3821657474a1b1e6b11ed6)
![{\displaystyle V_{0}={\frac {k_{2}[E]_{0}[S]}{K_{m}+[S]+K_{m}{\frac {[I]}{K_{i}}}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/34b2b90966cc2ce55ec35fde318bdcc2e984694b)
となる。
反応速度は全ての酵素が酵素-基質複合体として結合している時に最大となるため、
![{\displaystyle V_{\max }=k_{2}[E]_{0}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5d5602a3aa76c4c8e94949a07b8c7760704df376)
である。
項を置換し組み合わせると最終的に以下の慣習的な形式が得られる。
![{\displaystyle V_{0}={\frac {V_{\max }[S]}{K_{m}(1+{\frac {[I]}{K_{i}}})+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/27d69ec63802605dd919b1ce1dcdc80ecf98122c)
(8)
脚注
- ^ a b c d “Types of Inhibition”. NIH Center for Translational Therapeutics. 2012年4月2日閲覧。
- ^ “Competitive Inhibition”. 2012年4月2日閲覧。
- ^ “Enzyme Inhibitors”. 2014年9月4日閲覧。
- ^ “Enzyme Inhibition”. 2012年4月2日閲覧。
関連項目











